by Jonathan Latham, PhD and Allison Wilson, PhD
In China there is a popular joke about the southern city of Guangzhou (Canton). A visiting space alien, curious to learn about Chinese customs, tours its various provinces. Arriving in Guangzhou the alien asks the locals what their interests are. The Cantonese oblige their guest by putting the alien in a soup pot and eating it. This joke hinges on the Cantonese fondness for cooking with unusual species, many obtained from far away.
This feature of Canton’s cuisine was implicated in the original SARS (Severe Acquired Respiratory Syndrome) pandemic of 2002-04, which began in Guangzhou. It is thought that the virus arrived there with palm civets imported for speciality dishes (Wang et al., 2005).
But this culinary connection also marks a defining difference between the first SARS coronavirus pandemic and the current one. The COVID-19 (SARS-CoV-2) pandemic began in Wuhan, but Wuhan was considered a comparatively unlikely location for a natural (zoonotic) coronavirus spillover (Yu et al., 2019). It has no cultural or geographic or climatic predisposing factors.
For example, being fairly far north, bats are not abundant in Wuhan and Hubei province has few bat coronaviruses compared to hotspots like Yunnan and Guangdong (Yu et al., 2019). Unlike Canton, Wuhan is not famous for exotic fare. Nor is Wuhan near animal smuggling and trading origins (Li et al. 2019). It was for this reason that researchers from the Wuhan Institute of Virology (the WIV), which is the prime suspect in the various lab leak theories, mostly had to travel thousands of kilometres to find bats with coronaviruses (Yu et al., 2019). Furthermore, when WIV researchers needed to study a Chinese population that was not routinely exposed to bat coronaviruses (as a control group), they chose Wuhan residents (Wang et al. 2018; Li et al. 2019).
It is consequently a mystery, if SARS-CoV-2 does have a zoonotic origin, why COVID-19 should have emerged where it did. As Zheng-li Shi, head of coronavirus research at the WIV told Scientific American, in March 2020: “I had never expected this kind of thing to happen in Wuhan, in central China”.
What is the probability of a natural zoonotic coronavirus outbreak starting in Wuhan?
It is possible, and potentially helpful, to put numbers on Zheng-li Shi’s surprise. Numbers can more precisely show the incongruity of an outbreak occurring in Wuhan. But before using them it is important to specify the assumptions required so that these numbers can be treated with appropriate caution.
Such a calculation requires that we set aside momentarily all the varied, potentially important, but hard-to-quantify-and-mostly-unknown local factors, like those mentioned above, that may make certain locations or populations less or more likely to originate a pandemic. (For a broader discussion of these factors see e.g. Graham et al., 2013)
Given these proviso’s, and knowing that (1) bats and other animals which harbour coronaviruses are found practically all over the world, and (2) that the population of Wuhan is 11 million, and that (3) the global population is 7 billion, we can calculate the likelihood of Wuhan being the epicentre of a natural zoonotic coronavirus pandemic: The chance of a person from Wuhan being patient zero is approximately 1 in 630.
Therefore, if we were Zheng-li Shi, we would have “never expected” a natural zoonotic outbreak in Wuhan either. Imagine her surprise, and that of her colleagues when, in December 2019, they learned of a local coronavirus outbreak. They (and other researchers) travel all over the world, and not just China, looking for coronaviruses yet a pandemic breaks out in Wuhan, under their very noses. It truly is, very, very, unlikely that a natural zoonotic pandemic would start in Wuhan. Yet no commentator on the outbreak seems to have properly acknowledged the true scale of this improbability.
The second coincidence is an evolutionary coincidence
But there is, in fact, a second coincidence regarding the origin of the COVID19 pandemic. This coincidence has seemingly been entirely disregarded; but it too points strongly to a lab origin. The underlying logic is quite simple and it has to do with the evolution of coronaviruses.
Zheng-li Shi’s laboratory at the WIV is a world centre of coronavirus research. This has been mentioned often and is widely known. In particular, the Wuhan Institute of Virology is a world-leading site for bat coronavirus collection (and the virus came from a bat). But what has not been foregrounded is that, even within the coronaviruses, Zheng-li Shi’s laboratory had, of the 28 relevant coronavirus species, singled out just one of them as their special focus. And it is a member of this species (called the “SARS-related coronaviruses“) that broke out in Wuhan in 2019.
This, then, is a further curious coincidence: for a pandemic coronavirus (SARS-CoV-2) to emerge in Wuhan and be a member of the species most studied at the Wuhan Institute of Virology.
The logic of coronavirus pandemics
A fuller appreciation of this coincidence requires visualising coronavirus evolution and understanding the research agenda at the WIV.
The coronaviruses are divided into four types: Alpha-, Beta-, Gamma- and Delta- coronaviruses. These are shown in Fig. 1 which is a phylogenetic (evolutionary) tree adapted from a paper by Li et al., 2020. (The print is small and so here is a link to the original figure.)
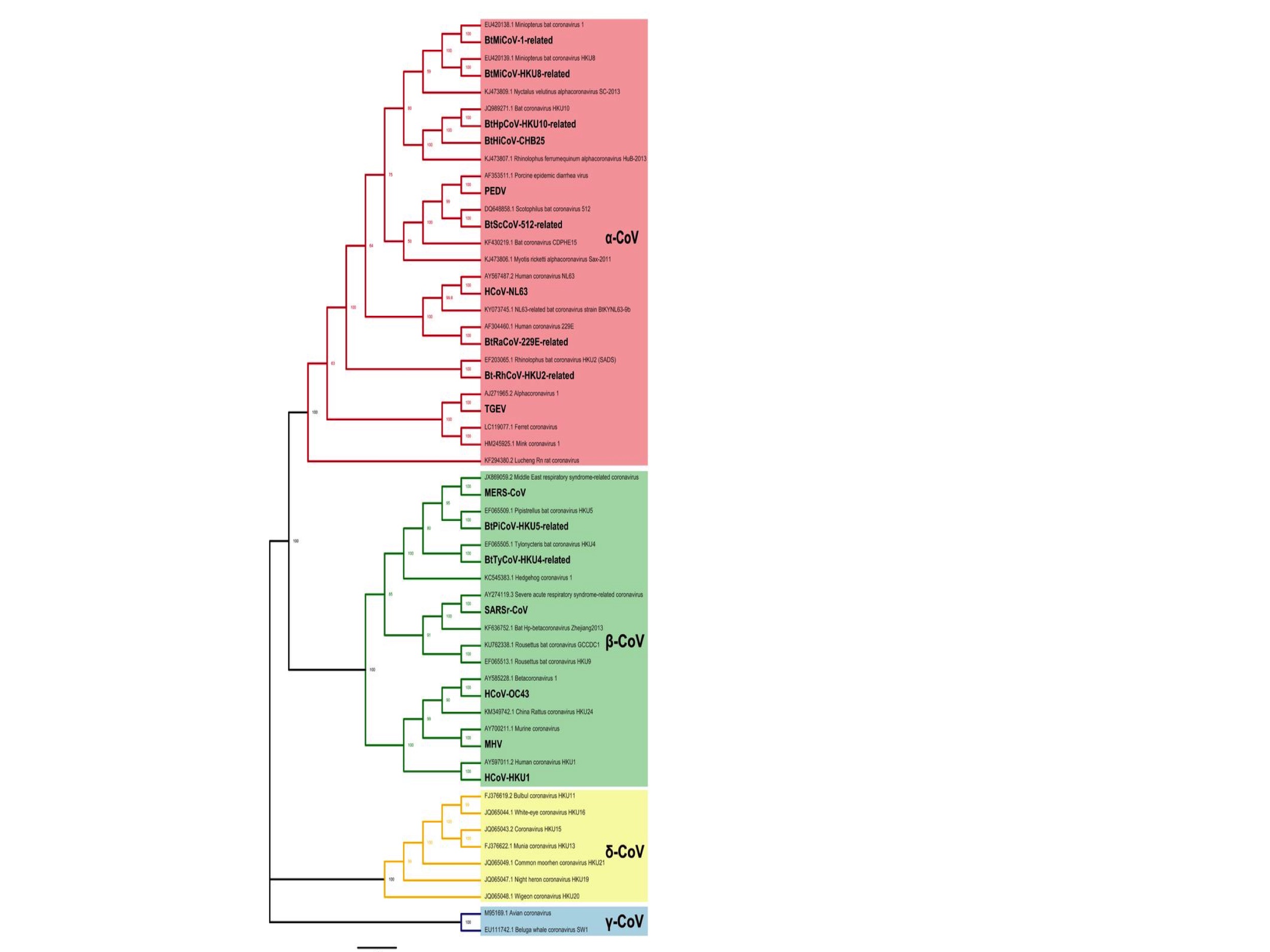
Of this phylogenetic tree, only the Alpha (pink) and Beta (green) coronaviruses will be considered here. This is because the Gamma (blue) and Delta (yellow) coronaviruses are few, not known to infect humans, and therefore questionably relevant.
As of February 2020, when Li et al. created this figure, there were 28 species of Alpha- and Betacoronaviruses. (Note: a species does not precisely equate to single tips on the phylogenetic tree in Fig 1. because some species have multiple members.)
It is important to appreciate, however, that we have no reason to suppose that a pandemic coronavirus could not have emerged from any branch of this phylogenetic tree. Indeed, the last coronavirus to jump into humans (before 2019) was MERS (Middle East Respiratory Syndrome) in 2012. MERS is a Betacoronavirus and was an unknown species before it started infecting humans. See the green arrow in Figure 2. The original SARS virus was also unknown as a species at the time it emerged as a human pathogen in 2002.
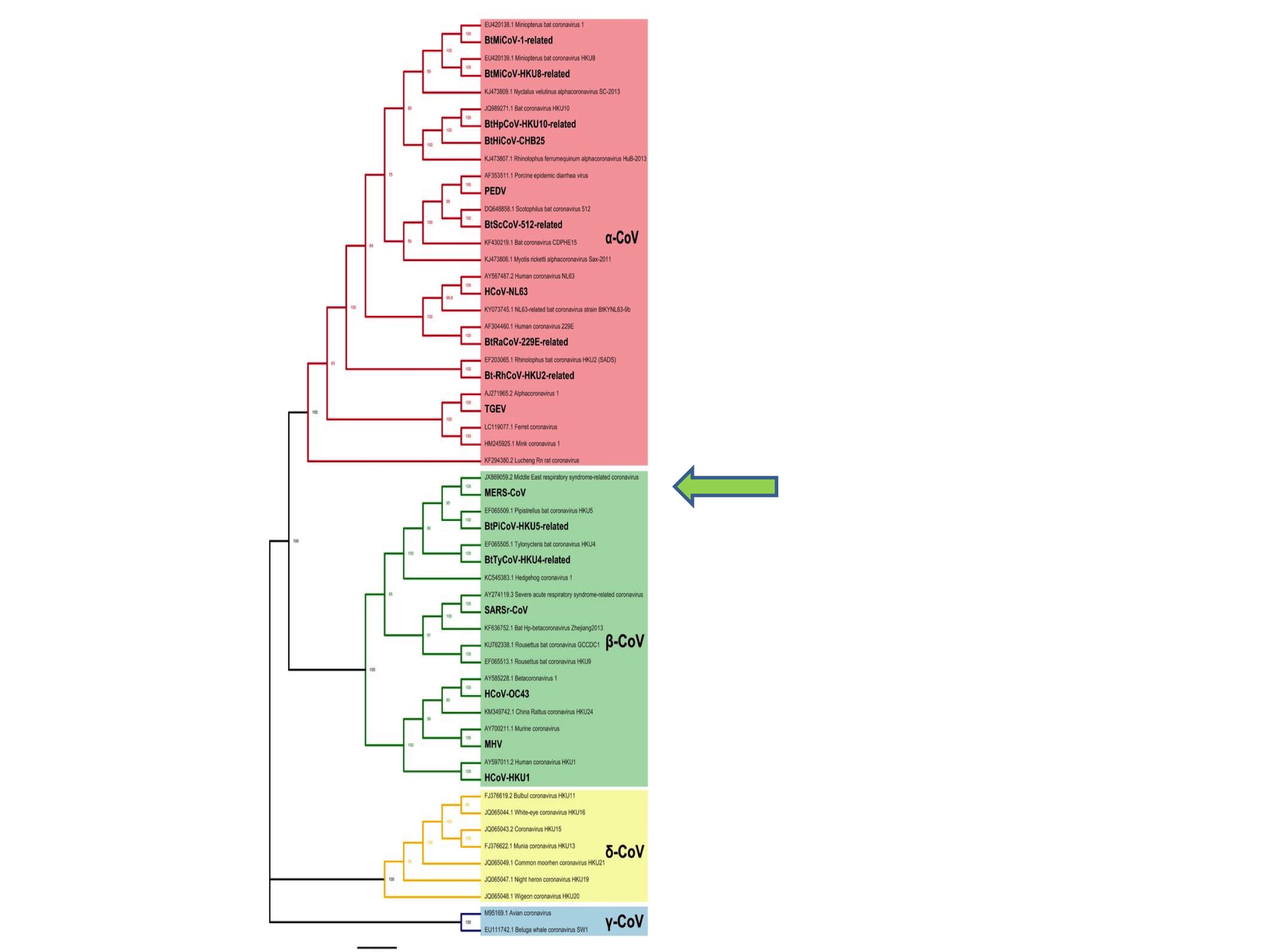
This unpredictability is also apparent from Zheng-li Shi’s choice of ‘disease X’. In 2018 the WHO announced a discussion list of pandemic priority diseases, which included Ebola, Rift Valley Fever, and other viruses. Alongside these known diseases the WHO asked experts to nominate a presently unknown candidate. Zheng-li Shi proposed that: “Disease X could be a transmissible infectious disease caused by a novel coronavirus originated from bats” (Jiang and Shi 2020). In other words, she did not predict any more narrowly than that the next pandemic would be caused by an Alpha- or Betacoronavirus.
The apparently random nature of coronavirus spillovers to humans is also apparent from inspection of Figure 3.
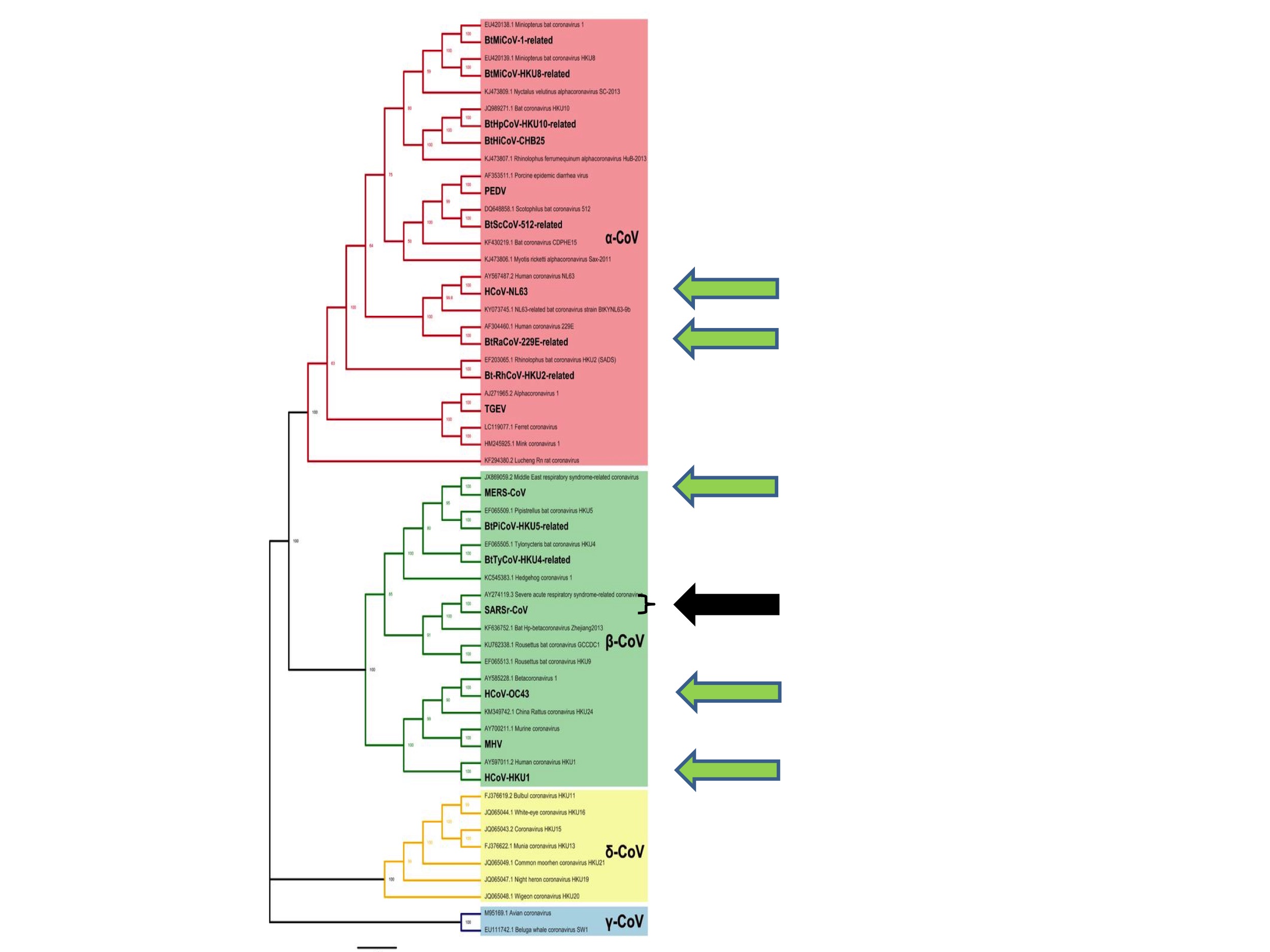
Figure 3 shows all of the six human coronaviruses identified prior to this pandemic. They are (from the top of the figure): HCoV-NL63, HCoV-229E, MERS, SARS, HCoV-OC43 and HCoV-HKU1. The six are each indicated in Figure 3 by green arrows, except for SARS, which is represented by a black arrow.
What Figure 3 illustrates is that human coronaviruses are distributed widely across the coronavirus family tree. That is to say, previous spillovers to humans happened at diverse and seemingly random points on the coronavirus tree and have involved both Alpha- and Betacoronaviruses.
The SARS-CoV-2 outbreak
With these prior assumptions stated we can then ask the question: where on the tree would one have expected (prior to the COVID-19 pandemic) the next novel coronavirus to emerge?
The answer is, if it were a natural or semi-natural spillover (i.e. a zoonosis)––from a random spot on the tree. It might have been an Alphacoronavirus or a Betacoronavirus. It might even, like MERS and SARS, be a novel species, since presumably there are still many undiscovered coronavirus species. The crucial point is that the chance of a spillover coming from each species is, as far as anyone knows, seemingly equal.
So where, phylogenetically speaking, did SARS-CoV-2 emerge?
The answer is shown in Figure 4 (below) in which the red arrow indicates the site of emergence of SARS-CoV-2.
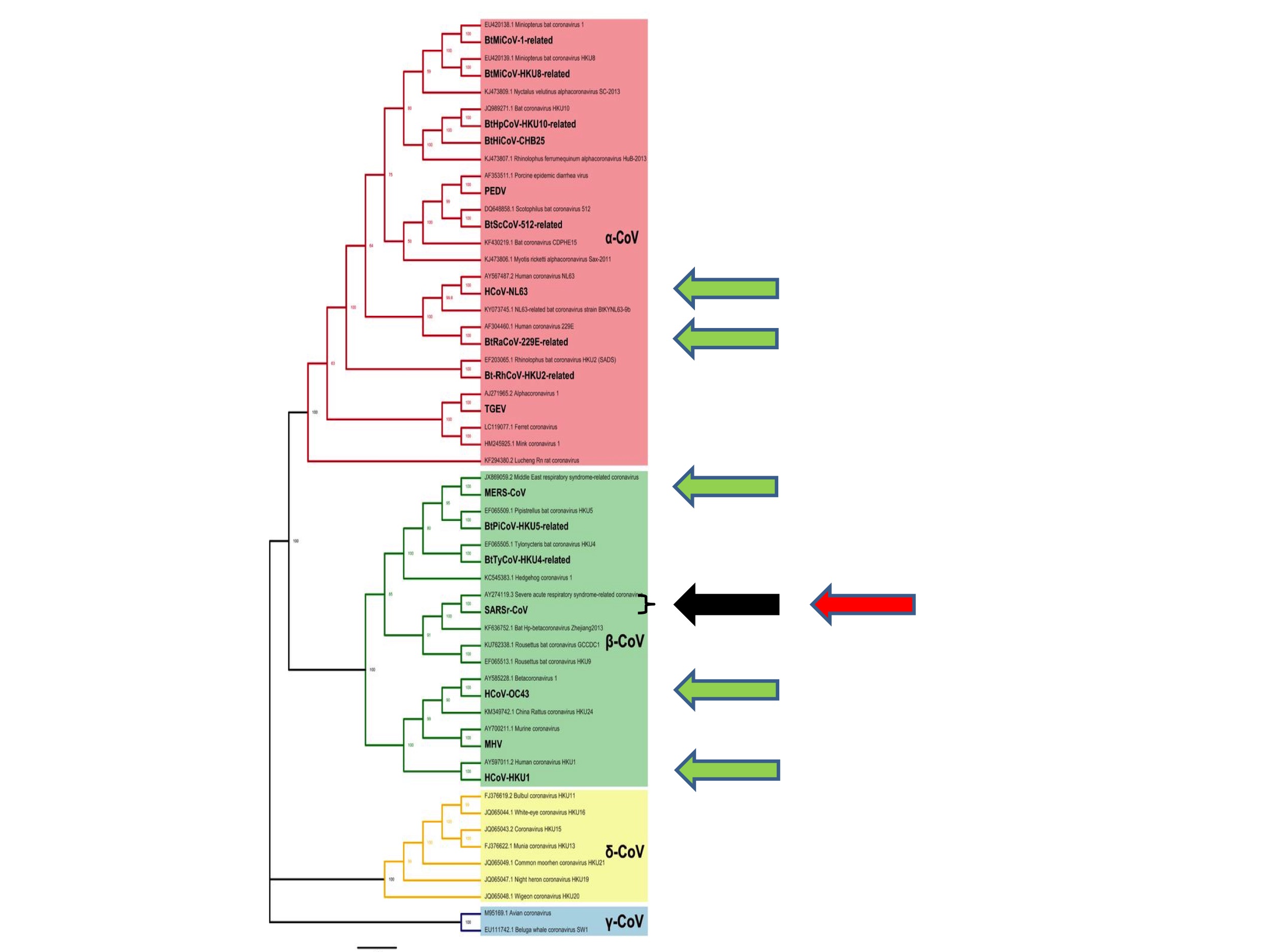
It emerged from the same species as the original SARS, hence its name. As noted above, this particular species is known to taxonomists as the “SARS-related coronaviruses” after its then most famous member (Coronavirus Study Group of the International Committee on Taxonomy of Viruses, 2020).
As discussed, from a zoonotic perspective, nothing appears to be special about these SARS-related coronaviruses. Consequently, the emergence of a second pandemic virus from the same coronavirus species constitutes a second surprising coincidence. We can again calculate its probability. If each Alpha and Betacoronavirus species is equally likely to spill over to humans, which is consistent with our understanding, then the probability of a virus from the SARS-related coronavirus species starting a zoonotic pandemic is 1 in 28. (And if there are undiscovered coronavirus species––pretty much a certainty––the number will be greater still).
It is a coincidence that, just like the emergence in Wuhan, heavily favours a lab escape if we take into account the specifics of the coronavirus research programme at the WIV, which are outlined below.
China’s research on SARS-related coronaviruses
Consider the following list of publication titles, many accepted in prestigious journals, from between 2005 and the start of the pandemic in late 2019. They are all authored by Zheng-li Shi. These eighteen research papers constitute the main focus of her published output. What they have in common is that all use the phrase “SARS-like coronavirus” or, later, “SARS-related coronavirus” or a close variant (all are bolded below). These phrases should be understood as technical terms. They denote viruses extremely closely related to SARS and only distantly related to other coronaviruses:
- ‘Bats Are Natural Reservoirs of SARS-like Coronaviruses‘ (2005);
- ‘Full-length genome sequences of two SARS-like coronaviruses in horseshoe bats and genetic variation analysis’ (2006);
- ‘Evidence of the recombinant origin of a bat severe acute respiratory syndrome (SARS)-like coronavirus and its implications on the direct ancestor of SARS coronavirus’ (2008);
- ‘Difference in Receptor Usage between Severe Acute Respiratory Syndrome (SARS) Coronavirus and SARS-Like Coronavirus of Bat Origin’ (2008);
- ‘Virus-like particles of SARS-like coronavirus formed by membrane proteins from different origins demonstrate stimulating activity in human dendritic cells’ (2008);
- ‘Immunogenicity difference between the SARS coronavirus and the bat SARS-like coronavirus spike (S) proteins’ (2009);
- ‘Intraspecies diversity of SARS-like coronaviruses in Rhinolophus sinicus and its implications for the origin of SARS coronaviruses in humans’ (2010);
- ‘Immunogenicity of the spike glycoprotein of Bat SARS-like coronavirus‘ (2010);
- ‘Bat severe acute respiratory syndrome-like coronavirus ORF3b homologues display different interferon antagonist activities’ (2012);
- ‘Identification of immunogenic determinants of the spike protein of SARS-like coronavirus‘ (2013);
- Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor (2013);
- ‘A SARS-like cluster of circulating bat coronaviruses shows potential for human emergence’ (2015);
- ‘Bat severe acute respiratory syndrome-like coronavirus WIV1 encodes an extra accessory protein, ORFX, involved in modulation of the host immune response’ (2016);
- Longitudinal surveillance of SARS-like coronaviruses in bats by quantitative real-time PCR’ (2016);
- ‘Cross-neutralization of SARS coronavirus-specific antibodies against bat SARS-like coronaviruses‘ (2017);
- ‘Discovery of a rich gene pool of bat SARS-related coronaviruses provides new insights into the origin of SARS coronavirus’ (2017);
- ‘Serological evidence of bat SARS-related coronavirus infection in humans, China’ (2018);
- ‘Geographical structure of bat SARS-related coronaviruses‘ (2019).
What this list demonstrates is that, while Zheng-li Shi at the WIV focused on virus collection, above all, she dedicated her research to understanding zoonotic spillovers to humans of one species alone: the SARS-related coronaviruses.
So while most discussions of a potential lab escape have mentioned that SARS-CoV-2 emerged within commuting distance of the WIV and that researchers at the WIV worked on bat coronaviruses, none have mentioned that the coincidence is much greater than that. Zheng-li Shi concentrated, especially with her potentially highly risky molecular research, on the particular species of coronavirus that is responsible for the pandemic.
There is a simple reason for this focus. The original SARS outbreak in 2002-04 had a major impact in China. Finding the origin, explaining SARS and its symptoms, and preventing a repeat all became major research priorities for Chinese scientists.
To be sure, Zheng-li Shi published papers on other coronavirus species over that same time-period, for example on MERS, and even some on non-coronaviruses; but these articles tended to be one-offs and co-authorships with other labs. The large majority of her output and the dominant theme of her research was collecting and manipulating SARS-related coronaviruses to determine the potential for human spillover.
So, if one accepts as reasonable the assumptions made above, the probability of Wuhan being the site of a natural SARS-related coronavirus outbreak is obtained by multiplying 1 in 630 by 1 in 28. The chance of Wuhan hosting a SARS-related coronavirus outbreak is thus 17,640–1.
The criticism will doubtless be made that the geographic and the phylogenetic evidence described here are circumstantial–mere coincidences. But critiquing evidence as circumstantial is based on a common logical misconception–that circumstantial evidence represents a special category of evidence. As the philosopher David Hume first argued, all evidence of causation is composed of coincidences. All an observer can do is to add up the coincidences until they surmise that the threshold of reasonable doubt has been surpassed. Conclusions are always provisional, but in the absence of evidence to the contrary, anyone open to persuasion ought at this point to conclude that a probability of 17,640–1 far exceeds that threshold. A lab escape should at this point be the default hypothesis.
Such a conclusion is only reinforced by much of the important information that has emerged since the outbreak began. We now know, for example, that, at the time of the outbreak, Zheng-li Shi and her colleagues had in their freezers the virus sample known as RaTG13. Among all the known coronaviruses, including within the SARS-related coronaviruses, RaTG13 is by far the closest relative of SARS-CoV-2. We also know that Zheng-li Shi implied she had not actively studied RaTG13 prior to the outbreak (in Zhou et al, 2020). We now know this was false and they had been studying it since at least 2017 (Zhou et al. 2020 addendum). These facts again do not support a natural zoonotic origin.
The lack of a zoonotic theory
If there were a credible zoonotic origin theory for the emergence of SARS-CoV-2 then such a calculation might be considered moot. But, despite considerable academic discussion (e.g. Leitner and Kumar, 2020; Seyran et al. 2020; Sallard et al., 2020) and a WHO investigation, there is still no substantive zoonotic theory to speak of. Snakes, Bamboo rats, pangolins, mink, turtles, dogs, civets, whales, and frozen cod, have all, at various times, been suggested as intermediate vectors that might have carried SARS-CoV-2, or coronavirus precursors of it, to Wuhan; but neither a theory, nor a proximal spillover virus, nor a plausible intermediate host has gained significant support in the scientific community. The excellent reason is that data supporting them are largely lacking despite the apparently very intensive searching (Sallard et al., 2020).
The most concrete of these zoonotic theories, and by far the most widely known, is the pangolin (Manis javanica) theory (Anderson et al., 2020; Lam et al., 2020; Xiao et al., 2020). It is proposed that pangolins smuggled from countries to the south of China harboured precursor coronaviruses picked up from bats, thereby bringing them to Wuhan.
However, newly available evidence has made this scenario improbable. First, pangolins do not seem, after all, to naturally carry coronaviruses (Lee et al., 2020). Second, the pangolin theory rests largely on virus sequences obtained from pangolins confiscated in Guangdong province in early 2019. Attempted independent verification of these virus sequences has uncovered that, although four publications (now highly cited) discuss or report pangolin coronavirus sequences and therefore appear to support the widespread presence of coronaviruses in pangolins, only one virus genome was ever sequenced (Chan and Zhan, 2020). The papers by Xiao et al. (2020) and Liu et al. (2020) merely renamed and reconfigured sequence information generated by Liu et al. 2019. This is the same pangolin coronavirus data set discussed by Lam et al., 2020. Current thinking, in light of this new evidence, is that the smuggled pangolins were an ‘incidental host’ of the coronavirus. That is, the pangolins likely caught the virus while being smuggled (Chan and Zhan, 2020; Lee et al, 2020).
In stark contrast, there are four distinct lab origin theories and these, unsurprisingly, are getting increasing attention. Two are published in the scientific literature (Sirotkin and Sirotkin, 2020; Segreto and Deigin, 2020). A third proposes that SARS-CoV-2 was a failed attempt to develop a vaccine. This theory was developed by an independent group of online researchers called DRASTIC. The fourth is our own Mojiang Miners Passage theory.
This latter theory starts from the fact that viruses in the same mine where RaTG13 (the closest related viral sequence to SARS-CoV-2) was sampled appear to have given rise to a disease outbreak in 2012. In that outbreak, six miners were hospitalized with COVID-19-like symptoms and three died (Rahalkar and Bahulikar, 2020). All had been shovelling bat guano and were diagnosed at the time as likely suffering from an unknown coronavirus. Samples from four of the hospitalized miners were sent to the WIV for testing. To-date, there are conflicting claims about the results of those tests and nothing has been formally published (Zhou et al. 2020 addendum). The Mojiang Miners Passage theory proposes, however, that, by the time they arrived at the WIV, these patient-derived samples contained a highly adapted human virus, which subsequently escaped.
For the present moment, notwithstanding the claim of the WHO investigation and the censorship of Facebook, all of these accidental lab origin theories appear plausible to us, but all remain uninvestigated. Our prediction, however, simply based on assessing the probabilities, is that no convincing natural zoonotic origin for the pandemic will ever be found by China or the WHO or anyone else––for the simple reason that one does not exist.
References
Andersen, K. G., Rambaut, A., Lipkin, W. I., Holmes, E. C., & Garry, R. F. (2020). The proximal origin of SARS-CoV-2. Nature medicine, 26(4), 450-452.
Chan, Y. A., & Zhan, S. H. (2020). Single source of pangolin CoVs with a near identical Spike RBD to SARS-CoV-2. BioRxiv.
S. G. of the International Committee on Taxonomy of Viruses (2020). The species Severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nature microbiology, 5(4), 536.
Graham, R. L., Donaldson, E. F., & Baric, R. S. (2013). A decade after SARS: strategies for controlling emerging coronaviruses. Nature Reviews Microbiology, 11(12), 836-848.
Jiang, S., & Shi, Z. L. (2020). The first disease X is caused by a highly transmissible acute respiratory syndrome coronavirus. Virologica Sinica, 35(3), 263-265.
Lam, T. T. Y., Jia, N., Zhang, Y. W., Shum, M. H. H., Jiang, J. F., Zhu, H. C., … & Cao, W. C. (2020). Identifying SARS-CoV-2-related coronaviruses in Malayan pangolins. Nature, 583(7815), 282-285.
Lee, J., Hughes, T., Lee, M. H., Field, H., Rovie-Ryan, J. J., Sitam, F. T., … & Daszak, P. (2020). No evidence of coronaviruses or other potentially zoonotic viruses in Sunda pangolins (Manis javanica) entering the wildlife trade via Malaysia. Ecohealth, 17(3), 406-418.
Leitner, T., & Kumar, S. (2020). Where did SARS-CoV-2 come from?. Molecular biology and evolution, 37(9), 2463-2464.
Li, H., Mendelsohn, E., Zong, C., Zhang, W., Hagan, E., Wang, N., … & Daszak, P. (2019). Human-animal interactions and bat coronavirus spillover potential among rural residents in Southern China. Biosafety and Health, 1(2), 84-90.
Li, B., Si, H. R., Zhu, Y., Yang, X. L., Anderson, D. E., Shi, Z. L., … & Zhou, P. (2020). Discovery of bat coronaviruses through surveillance and probe capture-based next-generation sequencing. Msphere, 5(1).
Liu, P., Chen, W., & Chen, J. P. (2019). Viral metagenomics revealed Sendai virus and coronavirus infection of Malayan pangolins (Manis javanica). Viruses, 11(11), 979.
Liu, P., Jiang, J. Z., Wan, X. F., Hua, Y., Li, L., Zhou, J., … & Chen, J. (2020). Are pangolins the intermediate host of the 2019 novel coronavirus (SARS-CoV-2)?. PLoS Pathogens, 16(5), e1008421.
Rahalkar, M. C., & Bahulikar, R. A. (2020). Lethal pneumonia cases in Mojiang miners (2012) and the mineshaft could provide important clues to the origin of SARS-CoV-2. Frontiers in public health, 8, 638.
Sallard, E., Halloy, J., Casane, D., Decroly, E., & van Helden, J. (2021). Tracing the origins of SARS-COV-2 in coronavirus phylogenies: a review. Environmental Chemistry Letters, 1-17.
Seyran, M., Pizzol, D., Adadi, P., El‐Aziz, T. M. A., Hassan, S. S., Soares, A., … & Brufsky, A. M. (2020). Questions concerning the proximal origin of SARS‐CoV‐2. Journal of Medical Virology.
Segreto, R., & Deigin, Y. (2020). The genetic structure of SARS‐CoV‐2 does not rule out a laboratory origin: SARS‐COV‐2 chimeric structure and furin cleavage site might be the result of genetic manipulation. BioEssays, 2000240.
Sirotkin, K., & Sirotkin, D. (2020). Might SARS‐CoV‐2 have arisen via serial passage through an animal host or cell culture? A potential explanation for much of the novel coronavirus’ distinctive genome. BioEssays, 42(10), 2000091.
Wang, M., Yan, M., Xu, H., Liang, W., Kan, B., Zheng, B., … & Xu, J. (2005). SARS-CoV infection in a restaurant from palm civet. Emerging infectious diseases, 11(12), 1860.
Wang, N., Li, S. Y., Yang, X. L., Huang, H. M., Zhang, Y. J., Guo, H., … & Shi, Z. L. (2018). Serological evidence of bat SARS-related coronavirus infection in humans, China. Virologica Sinica, 33(1), 104-107.
Xiao, K., Zhai, J., Feng, Y., Zhou, N., Zhang, X., Zou, J. J., … & Shen, Y. (2020). Isolation of SARS-CoV-2-related coronavirus from Malayan pangolins. Nature, 583(7815), 286-289.
Yu, P., Hu, B., Shi, Z. L., & Cui, J. (2019). Geographical structure of bat SARS-related coronaviruses. Infection, Genetics and Evolution, 69, 224-229.
Zhou, P., Yang, X. L., Wang, X. G., Hu, B., Zhang, L., Zhang, W., … & Shi, Z. L. (2020). A pneumonia outbreak associated with a new coronavirus of probable bat origin. nature, 579(7798), 270-273.




{kind=link}
January 26, 2024 @ 9:54 pm
Thanks for the ongoing updating on activity related to the Corona virus. This kind of attention and detail, at least for me, provides balance and a sense of security in this ongoing avalanche of information and helps me in my own inquiry. Arlene hickory